Genomics can transform precision health. We can detect cancer several years earlier through simple blood tests, without invasive biopsies. We can tailor treatment plans based on mutations in a cancer cell, and more.
Determining an individual’s DNA or whole-genome sequencing forms the foundation of genomics. Sequencing each genome (referred to as secondary analysis) could take hundreds to thousands of CPU hours depending on the read length. A genome is essentially a long string (3.08 Giga bp for a human genome) of DNA base-pairs (bp) A, G, C, and T. A sequencing machine splits a DNA into billions of small reads. In reference-guided assembly, these reads are aligned by matching them to a previously sequenced genome. This task is complicated by the fact that the new individual’s genome may not exactly match that of the reference genome. In fact, the end goal is to determine the variants in the new genome.
While there are several computational steps in sequencing raw genome data, we focus on accelerating read alignment, a time-consuming step in secondary analysis. To retain relevance in the medical community, it is critical that we adhere to the standard genomics analysis pipeline and not obtain efficiency gain at the cost of accuracy. Hence, unlike prior work, we ensure exact bit-level output equivalence to the Broad Institute’s community standard BWA-MEM pipeline.
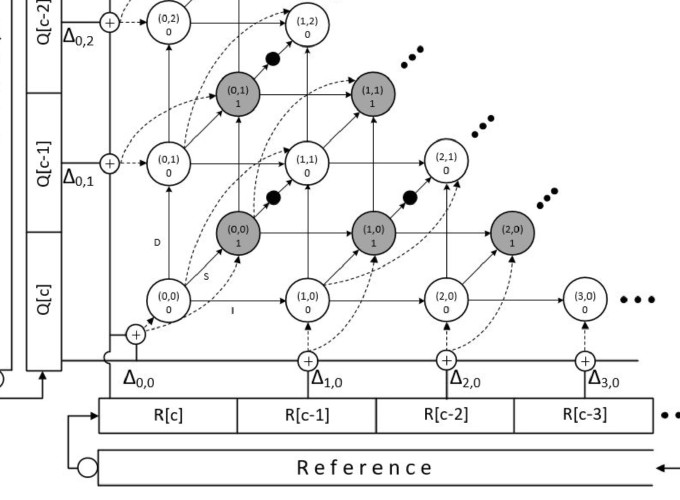
Read alignment determines the position of a read in the genome. Due to variants and sequencing errors, a read (referred as query Q) may not perfectly match a substring in the reference genome R. Sequence aligners solve this problem in two steps: seeding and seed-extension. Seeding finds perfect matches in the reference genome for small substrings (seeds) in a read. The seed positions are then extended to determine the best position by using an approximate string matching algorithm based on computing Levenshtein (edit) distance.
In this paper, we present GenAx, an ASIC custom hardware accelerator for both steps in read alignment. The seeding step is dominated by irregular memory accesses. We improve the locality of seeding by segmenting the genome to produce a smaller cache-able index for each segment. The seeds are then extended at high-throughput using SillaX, our proposed accelerator for approximate-string matching. SillaX is based on a novel automata design, Silla (String Independent Local Levenshtein Automata). Silla is string independent and its state space is only proportional to edit distance $K$, not string length $N$, unlike Levenshtein Automata (LA), which is string-dependent and needs $\mathcal{O}(KN)$ space. Its structure is also regular and composable, with only local communication between states. SillaX not only supports the edit distance scoring function, but also includes support for affine gap scoring and traceback, essential features required in sequencing.
GenAx provides 178$\times$ area-normalized alignment throughput compared to the state-of-the-art software (BWA-MEM) running on a 56-thread Xeon E5 processor. While performing read alignment on a single human genome using BWA-MEM takes about 5 hours and costs $\$$35 on an Amazon AWS instance with $\sim$56 processor cores, using GenAx, read alignment can be performed in $\sim$10 min. We also fabricated SillaX in the Mie Fujitsu 55 nm DDC technology as proof-of-concept.

